DNA METABARCODING INTRODUCTION
DNA METABARCODING MANUAL SEQUENCE EDITING
The files output by an Illumina MiSeq are in FASTQ format. This is similar to the FASTA which you are familiar with but contains additional data. We get two FASTQ files from each sequencing run, which you can think of as the Forward and Reverse sequences.
Some example FASTQ Paired-end are available here. For convenience, save them in a folder on your Desktop. Follow the steps below to edit these DNA metabarcodes.
For the first step we use the online version of the program PRINSEQ.
a. Click on the image below to go to the page, and click on Upload Data.
Some example FASTQ Paired-end are available here. For convenience, save them in a folder on your Desktop. Follow the steps below to edit these DNA metabarcodes.
For the first step we use the online version of the program PRINSEQ.
a. Click on the image below to go to the page, and click on Upload Data.
b. The example files are FASTQ Paired-end so choose that option.
c. Select the two FASTQ files you have saved on your Desktop.
d. Under Please select the statistics you want to generate. choose None for all options then click Continue.
e. Wait while PRINSEQ processes your data.
f. Once its done click Process Input Data.
g. Choose the following options.
c. Select the two FASTQ files you have saved on your Desktop.
d. Under Please select the statistics you want to generate. choose None for all options then click Continue.
e. Wait while PRINSEQ processes your data.
f. Once its done click Process Input Data.
g. Choose the following options.

h. Choose to Output the data as FASTA, Data passing all the filters (good).
i. Click Generate Files.
j. Use right click on the file you want (all of the FASTA files) to download and select "Save Link As" to save the file.
Next we will be using the CodonCode Aligner program again.
k. Import the sequences in the two FASTA files. Click Rename Duplicates.
l. Select all the sequences and assemble based on 97.5% sequence identity. Make sure the settings for Assembly found under Edit>Preferences are set as follows.
i. Click Generate Files.
j. Use right click on the file you want (all of the FASTA files) to download and select "Save Link As" to save the file.
Next we will be using the CodonCode Aligner program again.
k. Import the sequences in the two FASTA files. Click Rename Duplicates.
l. Select all the sequences and assemble based on 97.5% sequence identity. Make sure the settings for Assembly found under Edit>Preferences are set as follows.
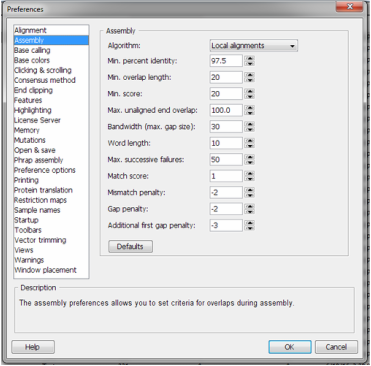
m. Select all the contigs, including the Unassembled Samples folder and again, assemble based on 97.5% sequence identity, repeat until cannot reduce the number of contigs anymore.
n. Delete (Move to Trash) the sequences still remaining in the Unassembled Samples folder.
o. Export all the remaining contigs as consensus sequences (FASTA file). Make sure none of the Options are selected.
p. Import the FASTA file into CodonCode (new project).
q. Assemble the sequences at 79% sequence identity and delete (Move to Trash) any sequences that don’t form into the main contig.
r. Unassemble then Assemble at 80%-95% (whichever gives a reasonable number of contigs) and check each contig carefully. We choose 90% because we have found that chimeras are usually within 10% of one of their parent sequences. If there are multiple sequences with slight divergences and they are different length, e.g. one short and one long, you can delete the short one. The presence of gaps, especially long ones, may indicate chimeras.
s. Please repeat step r as necessary until your happy with the quality of remaining sequences.
t. Unassemble then Assemble at 95%.
u. Switch bases to amino acids, check each reading frame and delete any singletons/contigs with stop codons. Double click the sequence, click the button GAT>ASP at the top of the window to change bases to amino acids or vice versa and click 123 to change the reading frame. Make sure CodoncodeAligner is set to the correct genetic code (Click Edit, Preferences, change the Organism accordingly, for insects will be Drosophila mitochondria. Make sure the contigs/sequences start from 5’ end to 3’end (For example: if your sequence starts from 3’ end to 5’ end and shows some stop codons, stop codons might not appear in your sequence if you reverse complement your sequence to 5’ end to 3’ end. You might remove good sequences if your sequences did not start from 5’ end to 3’ end).
v. Export the consensus sequences and individual sequences as FASTA files.
w. These are your final OTU for taxonomy matching.
x. You can align these sequences in BIOEDIT (see 10. SEQUENCE ALIGNMENT) and upload them to BOLD (see 12.SEQUENCE AND TRACE UPLOAD TO BOLD).
Those interested in learning more about DNA metabarcoding can consider enrolling in a Metabarcoding Spring School, now in it 6th edition.
n. Delete (Move to Trash) the sequences still remaining in the Unassembled Samples folder.
o. Export all the remaining contigs as consensus sequences (FASTA file). Make sure none of the Options are selected.
p. Import the FASTA file into CodonCode (new project).
q. Assemble the sequences at 79% sequence identity and delete (Move to Trash) any sequences that don’t form into the main contig.
r. Unassemble then Assemble at 80%-95% (whichever gives a reasonable number of contigs) and check each contig carefully. We choose 90% because we have found that chimeras are usually within 10% of one of their parent sequences. If there are multiple sequences with slight divergences and they are different length, e.g. one short and one long, you can delete the short one. The presence of gaps, especially long ones, may indicate chimeras.
s. Please repeat step r as necessary until your happy with the quality of remaining sequences.
t. Unassemble then Assemble at 95%.
u. Switch bases to amino acids, check each reading frame and delete any singletons/contigs with stop codons. Double click the sequence, click the button GAT>ASP at the top of the window to change bases to amino acids or vice versa and click 123 to change the reading frame. Make sure CodoncodeAligner is set to the correct genetic code (Click Edit, Preferences, change the Organism accordingly, for insects will be Drosophila mitochondria. Make sure the contigs/sequences start from 5’ end to 3’end (For example: if your sequence starts from 3’ end to 5’ end and shows some stop codons, stop codons might not appear in your sequence if you reverse complement your sequence to 5’ end to 3’ end. You might remove good sequences if your sequences did not start from 5’ end to 3’ end).
v. Export the consensus sequences and individual sequences as FASTA files.
w. These are your final OTU for taxonomy matching.
x. You can align these sequences in BIOEDIT (see 10. SEQUENCE ALIGNMENT) and upload them to BOLD (see 12.SEQUENCE AND TRACE UPLOAD TO BOLD).
Those interested in learning more about DNA metabarcoding can consider enrolling in a Metabarcoding Spring School, now in it 6th edition.
